Kapitel: Venöse Malformationen
Artikel: 12 von 13
Update: 2020/01/28
Autor/en: Barbera, Letterio Christian
Hierzu zählen die segmentalen Überwuchssyndrome sowie das Servelle-Martorell- und das Maffucci-Syndrom. Die Patienten weisen neben venösen Malformationen eine umschriebene und asymmetrische Entwicklung von Bindegewebe und Skelettsystem auf.
Proteus-Syndrom, CLOVES-Syndrom und Klippel-Trénaunay-Syndrom sind gekennzeichnet durch eine somatische Mutation, die nicht vererbt wird und nur im betroffenen Körperabschnitt nachweisbar ist (genetisches Mosaik). Wie im Namen definiert, liegt ein partieller Riesenwuchs vor, der meistens eine Körperhälfte (Gesicht, Extremität) betrifft. Umfangs- als auch Längenunterschiede variieren während des Wachstums, wobei deren Ausmaß nicht vorhergesagt werden kann. Eingehende Untersuchungen zum natürlichen Verlauf liegen nicht vor, der Eindruck besteht allerdings, dass der Überwuchs in der ersten Wachstumsphase (2–4 Lj) ausgeprägter als in der dritten (11–14 Lj) ist.
Der asymmetrische Knochenwuchs wird erst nach der Geburt beobachtet, nimmt während der Wachstumsphasen stark überproportional zu und kann umschrieben Schädel, Stamm und Extremitäten betreffen. Die Größenverhältnisse des Knochens können derart verändert sein, dass die ursprüngliche Form nicht mehr erkenntlich ist. An den betroffenen Körperabschnitten kann die Subkutisdicke sowohl stärker als auch schwächer als an der kontralateralen Körperseite ausgebildet sein. Gutartige Hauttumoren mit hirnwindungsähnlicher Oberfläche sind für dieses Syndrom pathognomonisch (sogenannter Cerebriform Mixed Connective Tissue Nevus). Die assoziierten Gefäßfehlbildungen treten in aller Regel als kombinierte slow-flow-Malformationen (LVM, CVM, CLVM) auf.
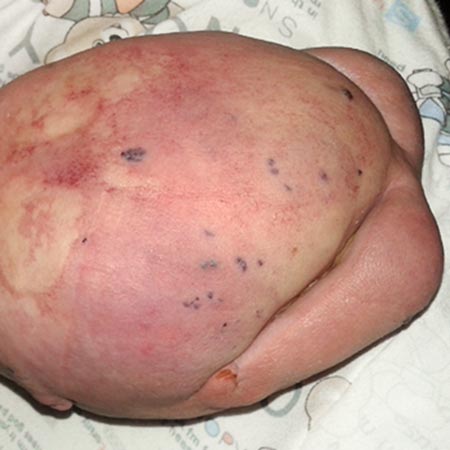
Umschriebene Fettgewebsvermehrung, kongenitale Gefäßfehlbildungen (VM, CM, LM und AVM) und epidermale Naevi, die an Kopf, Stamm und Extremitäten auftreten können, kennzeichnen dieses Großwuchssyndrom. Trotz der Gutartigkeit des Leidens, bedingt die auffällige Entstellung des betroffenen Körperabschnittes eine relevante Beeinträchtigung der Lebensqualität, die teilweise durch plastisch-chirurgische Eingriffe korrigiert werden kann.
Das Klippel-Trénaunay-Syndrom (KTS) im engeren Sinne ist die zuerst beschriebene angeborene Gefäßfehlbildung, wobei der Begriff häufig fälschlicherweise für andere vaskuläre Malformationen verwendet wird.
Kennzeichnend für das KTS ist die Trias: Dysproportionaler Überwuchs einer Extremität bzw. Körperhälfte + Kapilläre Malformation (Naevus flammeus) + Venöse Malformation +/- Lymphatische Malformation. Der Riesenwuchs kann erhebliche Ausmaße erreichen und schwerste Gehbehinderungen bedingen. Somit stellt es eine Anomalie dar, die das Leben des Betroffenen stark beeinträchtigt.
Die im Kindesalter auffälligeren Gefäßektasien spielen eine klinisch weniger relevante Rolle. Entgegen früherer Annahmen sind die fehlgebildeten Gefäße nicht für den Überwuchs verantwortlich sondern begleitende primäre Dysplasien des Weichteilgewebes. Die vaskulären Anomalien erstrecken sich über die betroffene Körperhälfte mit Prädilektion der lateralen Anteile. Während die kapilläre Malformation in aller Regel abblasst und manchmal nur an den sekundär als Abflussgefäße entstandenen retikulären Venen noch erkennbar ist, so nimmt die VM im Laufe der Jahre zu. Kaliberstarke, an der Außenseite des Beines subkutan verlaufende Gefäße münden in Höhe von Knie, Oberschenkel oder gluteal in das tiefe Venensystem ein. Wenngleich diese venöse Malformation als Marginalvene bzw. Embryonalvene bei Aplasie der Leitvenen bezeichnet wird, so entspricht die vorliegende Gefäßmorphologie oft eher einem Venengeflecht als einem singulären Gefäß.
Diese Erkenntnisse sind für Therapieplanung und Prognose relevant. Die Indikation für eine frühzeitige, ausgedehnte Resektion der venösen Malformation ist somit kritisch zu überprüfen. Eine konsequente Kompressions- und Bewegungstherapie mit selektiver Behandlung schmerzhafter Areale erscheint die bessere Alternative zu sein. Kommunikationen mit dem tiefen Leitvenensystem sind zu verschließen.
Das PTEN-Hamartoma-Tumor-Syndrom (PTEN-HTS) umfasst eine heterogene Gruppe seltener Erkrankungen mit partiellem Riesenwuchs, Weichteiltumoren und slow-flow- sowie fast-flow-Gefäßmalformationen unklaren Verhaltens. Die Neubildungen können an Haut, Schleimhäuten, Brustdrüse und Darmtrakt vorkommen und gelten als Präkanzerosen. Gemeinsames Merkmal dieser Syndromgruppe ist das Vorliegen einer autosomal-dominanten Mutation des PTEN-Gens, das im Blut nachweisbar ist. Die assoziierte VM spielt im Gegensatz zur oft begleitenden AVM eine klinisch weniger relevante Rolle. Wichtig ist daran zu denken, dass ein solches Syndrom vorliegen kann, wenn venöse Malformationsanteile in besonders große, meist fetthaltige Weichteilgewebsmassen eingebettet sind.
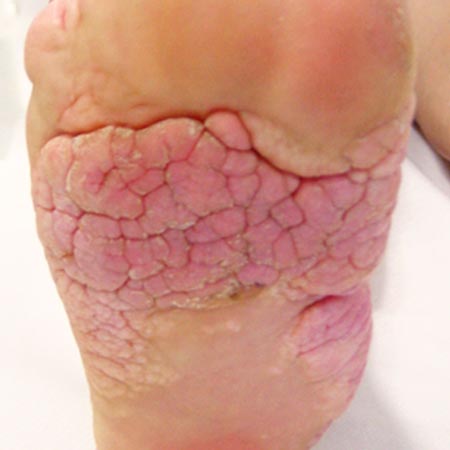
Auch „Blue rubber bleb nevus syndrome“ (BRBNS) genannt, ist das Bean-Syndrom eine selten auftretende sporadische Erkrankung, die venöse Malformationen an Haut, Schleimhäuten und inneren Organen aufweist. Die multifokal auftretenden Läsionen sind livide bis dunkelblau verfärbt, leicht erhaben und oft nur wenige mm groß. Die Oberfläche hat eine gummiartige Konsistenz, blasst unter Druck ab und ist selten schmerzhaft. Die im Kindesalter auftretende kutane Manifestation ist bis auf das Aussehen klinisch nicht relevant. Später treten diese Gefäßveränderungen auch an den inneren Organen auf und werden durch wiederkehrende Blutungen auffällig. Am häufigsten ist der Gastrointestinaltrakt (GIT) betroffen, wo sowohl akute als auch chronische Blutverluste auftreten. Diese manifestieren sich wie jede andere GIT-Blutung (Hämatemesis, Meläna, peranale Blutabgänge) und können lebensbedrohlich werden. Der chronische Blutverlust führt entsprechend zur Eisenmangelanämie und Abgeschlagenheit. Die Behandlung besteht in Bluttransfusionen, Eisensubstitution, endoskopischer Blutstillung, oft mit endoskopisch gesteuerter Sklerosierung und, falls diese nicht möglich ist, in Teilresektionen des betroffenen Darmabschnittes. Kontrollendoskopien sind nach Diagnosestellung die wichtigste Vorsorgemaßnahme. Weitere, wenngleich seltenere, extrakutane Lokalisationen sind in Knochen, Gehirn und Unterleib, wo Hirnblutungen, pathologische Frakturen, schwere Menorrhagien auftreten können. Während des letzten Trimenon nimmt das Volumen der Fehlbildungen zu, sodass eine erhöhte Blutungsgefahr unter der Geburt besteht. Eine entsprechende multidisziplinäre Betreuung der Schwangeren ist dann notwendig.
Bei dieser Sonderform tritt die venöse Malformation zusammen mit einem partiellen Minderwuchs einer Extremität auf. Die Genese des Syndroms ist unbekannt. Auffällig sind Umfangsunterschiede wofür sowohl eine Hypoplasie des Unterhautfett- als auch des Muskelgewebes verantwortlich sind. Geringe Längenunterschiede der Röhrenknochen bedingen eine Minusdifferenz und sind orthopädisch-chirurgisch nicht auszugleichen. Eingriffe zu Verlangsamung des Längenwachstums am kontralateralen Bein sind meist nicht indiziert. Die Behandlungsmöglichkeiten wie körperliches Training und Schuhhöhenausgleich sind symptomatischer Natur, sollten dennoch konsequent ausgeschöpft werden. In ausgewählten Fällen können Interventionen notwendig werden.
Diese seltene Erkrankung mit Knochen- und Gefäßanomalien, die häufig die obere Extremität betrifft, ist mit einer heterozygoten, somatischen Mutation assoziiert. Das Manifestationsalter liegt bei 4–5 Jahren und der Verlauf ist in aller Regel symptomarm. Neben großlumigen, venösen Malformationen treten multiple Enchondrome auf, die Achsendeformitäten und auffällige Auftreibungen an Röhrenknochen zur Folge haben. Diese Enchondrome können im Langzeitverlauf entarten und müssen kontrolliert werden. Pathologische Frakturen können auftreten, sodass entsprechende Vorsichtsmaßnahmen empfohlen sind. Die Behandlung richtet sich am Beschwerdebild, wobei ästhetische Korrektureingriffe wegen erhöhter Komplikationsraten (Fraktur, Blutung) einer strengen Indikationsstellung bedürfen.