Kapitel: Gefäßmalformationen assoziiert mit anderen Anomalien
Artikel: 11 von 13
Update: 2021/01/15
Autor/en: Tinschert, Sigrid | Wohlgemuth, Walter A.
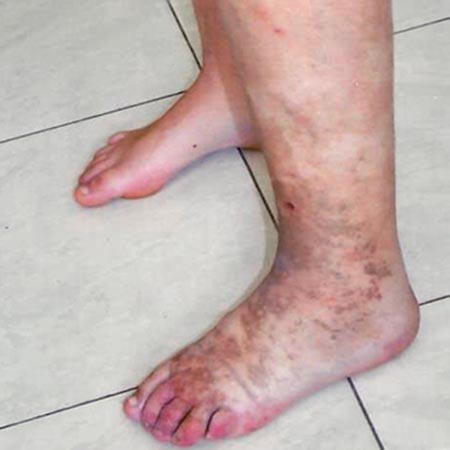
Das Parkes Weber-Syndrom (PWS) ist charakterisiert durch die Kombination von kutanen kapillären Malformationen (KM) und arteriovenösen Malformationen (AVM) bzw. multiplen im Gewebe liegenden, sehr feinen arteriovenösen Fisteln (AVF) mit einem ipsilateralen umschriebenen Überwuchs einer Extremität. Lymphatische Malformationen (LM) können ebenfalls vorliegen und treten als Ödem in Erscheinung.
Das Parkes Weber-Syndrom tritt meist sporadisch (nicht familiär) auf, selten ist ein familiäres Vorkommen (Paradominanz). Ursächlich sind Funktionsverlust-Mutationen im RASA1-Gen, das für das p120-Ras-GTPase aktivierende Protein (p120-RasGAP) kodiert und aktives, an GTP-gebundenes Ras, in die inaktive, GDP-gebundene Form überführt. Eine Gefäßmalformation entsteht vermutlich erst durch den kompletten Funktionsverlust von RASA1 infolge biallelischer Mutationen (LOH = „Verlust der Heterozygotie“). Die segmentalen Veränderungen (lokalisierte vaskuläre Malformationen und Überwuchs) wären durch einen frühembryonalen LOH des RASA1-Gens vor dem Hintergrund einer ererbten konstitutionellen (in allen Zellen vorliegenden) RASA1-Mutation erklärbar und entsprechen einem sog. Typ-2-Mosaik (nach Happle: Syndrom des Typ 2 segmentalen Naevus rhodoides). Multiple CM/AVM ohne Überwuchssymptomatik stellen eine oligosymptomatische Variante dar und sind erklärbar durch spätere (nicht-frühembryonale) RASA1-Mutationen.
Der regionale Überwuchs (Subkutanfettgewebe, Muskulatur und Knochen) betrifft meist eine Extremität oder Teile davon. Die Beine sind häufiger betroffen als die Arme.
Die kapillären Malformationen (KM) und kleine, in der Subkutis gelegene arteriovenöse Kurzschlüsse (Mikro-AVM/-AVF) führen zum typischen Bild der rosa- bis rotfarbenen (rhodoiden) Nävi des PWS. Die Nävi sind von variabler Größe und Zahl, wobei zumindest immer ein großer Nävus vorhanden ist. Kleinere Nävi sind gehäuft mit einem Halo umgeben. Die betroffene Hautregion ist durch die shuntbedingte Mehrperfusion überwärmt.
Mikrofistulöse-AVM/-AVF sind außer in der Subkutis ausgedehnt auch in der Muskulatur und im Knochen zu finden.
Lymphatische Malformationen können zusätzlich vorkommen und äußern sich als Lymphödem.