Kapitel: HHT · Morbus Osler
Artikel: 2 von 13
Update: 2020/06/20
Autor/en: Tinschert, Sigrid | Kühnel, Thomas
Es handelt sich um eine autosomal-dominant erbliche Krankheit. Nach aktuellem Kenntnisstand sind Mutationen in drei verschiedenen Genen ursächlich. Mutationen im ENG (Endoglin)-Gen (auf Chromosom 9), haben den HHT Typ 1 (ca. 25–60 %) zur Folge, Mutationen im ACVRL1-Gen (auf Chromosom 12), führen zum HHT Typ 2 (40–60 %). Sehr selten sind Mutationen im GDF2/BMP9-Gen (auf Chromosom 10), die zum HHT Typ 5 führen. Die Existenz zweier weiterer Typen, HHT Typ 3 und HHT Typ 4, basiert nur auf genetischen Kopplungsuntersuchungen; krankheitsursächliche Gene wurden dafür noch nicht identifiziert. Eine Sonderform stellt die Kombination von hämorrhagischen Teleangiektasien (HT) mit juveniler Polypose (JP) dar. Diese Krankheit (JPHT) basiert auf Mutationen des Tumorsuppressor-Gens SMAD4 (auf Chromosom 18) und betrifft ca. 1 bis 2 % der Patienten mit HHT.
Alle involvierten Gene kodieren für Proteine, die im TGF-beta (ß)-Signalweg eine Bedeutung haben. Von diesem Signalweg gehen eine Reihe von zellulären Funktionen der Angiogenese aus. Die Proteine werden in den Endothelzellen exprimiert.
Mit der Untersuchung der Gene ENG, ACVRL1 und SMAD4 kann bei ca. bei 75 % der Personen mit HHT die genetische Ursache nachgewiesen werden.
Wenn die Mutation in einer HHT-Familie bekannt ist, kann gezielt nach Merkmalsträgern auch unter noch asymptomatischen Personen in dieser Familie gefahndet werden. Damit ist eine Chance eröffnet, klinisch inapperente aber doch komplikationsträchtige pAVM (pulmonale arteriovenöse Malformationen) und cAVM (cerebrale arteriovenöse Malformationen) zu diagnostizieren. Eine solche prädiktive Diagnostik darf (laut Gendiagnostikgesetz) nur im Rahmen einer genetischen Beratung durch einen Facharzt für Humangenetik veranlasst werden.
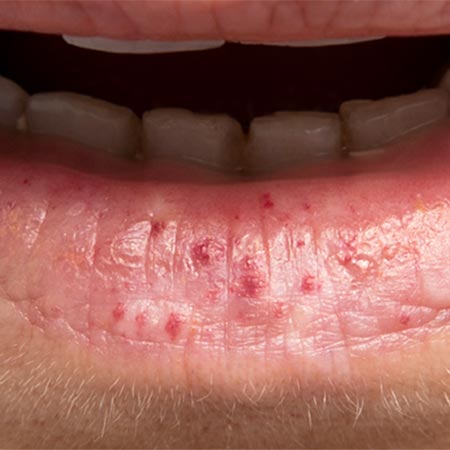
Es besteht eine gewisse Genotyp-Phänotyp-Beziehung: Bei HHT Typ1 (ENG) gibt es eine höhere Prävalenz von pulmonalen AVM (pAVM) und zerebralen vaskulären Malformationen (CVM) als bei HHT Typ 2. Bei HHT Typ 2 (ACVRL-Mutationen) wiederum liegen häufiger vaskuläre Malformationen der Leber und pulmonale Hypertonie vor. Bei der Sonderform JPHT (SMAD4) steht klinisch meist eine Polypose im Vordergrund. Von den HHT-Merkmalen sind Epistaxis (kutane Teleangiektasien können fehlen) und pAVM am häufigsten.
Die Entstehung der vaskulären Läsionen an bestimmten Stellen der Nasenschleimhaut ist durch den in allen Körperzellen vorhandenen Gendefekt (Keimbahnmutation) alleine nicht erklärbar. Für die Bildung der letztlich blutenden Gefäßveränderungen bedarf es zusätzlich zur Keimbahnmutation eines weiteren Schrittes, des sogenannten „second hit“ in den Endothelzellen. Kürzlich konnte gezeigt werden, dass es sich bei dem „second hit“ um ein Mutationsereignis handeln kann, welches das zweite Allel des entsprechenden Gens betrifft. Die Folge ist ein biallelischer Funktionsverlust des Proteinproduktes in den betroffenen Endothelzellen. Ob auch andere angiogene Trigger als „second hits“ fungieren können, wie lange Zeit vermutet wurde, ist noch nicht abschließend geklärt.